- Purpose: defines the prefix/suffix for the files (including path to the directory) containing real part of the electronic Hamiltonian matrices for all time steps of the simulation (their range is determined by namdtime parameter and the initial conditions). Note that the real part of the Hamiltonian is typically close to zero for the off-diagonal element, while the diagonal elements contain eigenenergies of corresponding KS orbitals. The format of the file content is simple - N x N matrix with real (floating-point values) entries, where N - is the number of KS orbitals considered in calculations (see more details in ...)
- Value type: string
- Possible values:anything
- Default value: ""
- Purpose: similarly to Ham_re_prefix / Ham_re_prefix, but for imaginary part of the Hamiltonian matrix. Typically the diagonal
terms of such matrix are zero, while the off-diagonal terms contain -i*hbar*
- Value type: string
- Possible values: anything
- Default value: ""
- Purpose: defines the prefix/suffix for the files (including path to the directory) containing x-component of the transition dipole matrix elements for all time steps of the simulation (their range is determined by namdtime parameter and the initial conditions). These files are required is explicit field-matter interaction is to be included in simulations or if one wishes to compute absorption spectra
- Value type: string
- Possible values: anything
- Default value: ""
- Purpose: similarly to Hprime_x_prefix / Hprime_x_suffix, but for y-component
- Value type: string
- Possible values: anything
- Default value: ""
- Purpose: similarly to Hprime_x_prefix / Hprime_x_suffix, but for z-component
- Value type: string
- Possible values: anything
- Default value: ""
- Purpose: Defines the type of calculations you want to perform
- Value type: string
- Possible values:
"namd" - perform NA-MD calculations (with or without decoherence, field, etc.)
"nonamd"(or anything else) - only calculate the energies and input states and parameters. No trajectory hopping and TD-SE calculations are performed, no output is written (except for me_energies*). This is helpful for computing the energies of the excited states (determinants) along the trajectory.
- Default value: "namd"
- Purpose: Determined how (at which stage) the Hamiltonian (and, optionally, Hprime) matrices are read.
- Value type: string
- Possible values:
"batch" - in this case all Ham_ (and, optionally, Hprime_) files (as defined by namdtime and initial conditions) are read first and stored internally. This helps to save a great deal of time, because reading this (same) files for each initial condition is very time consuming part. The only drawback of this method is that a lot of memory may be required. This option is preferred if the memory allows. Also note that this mode requires ALL Ham_ (and, optionally, Hprime_) files to be read in the beginning, even if the lowest initial excitation time is much larger than 0. So such files must be present.
"online" - in this case Ham_ (and, optionally, Hprime_) files are read as they need. This may be much slower than batch mode, but requires much less memory, because only current file is stored. This mode requires only those files which are actually used - that may be convenient in some cases.
"online_all_in_one" - this is similar to "online", but now both imaginary and real part are combined in a single Hamiltonian file. See example of usage and more explanation here
- Default value: "online"
- Purpose: Defines the length of the simulation, in number of steps (whatever the size of each MD step) - usually the size of such step is 1 fs.
- Value type: integer
- Possible values: any, starting from 0
- Default value: 0
- Purpose: Number of surface-hopping trajectories (independent) calculated for each initial condition. The output populations are averaged over this number of trajectories.
- Value type: integer
- Possible values: any bigger than 0
- Default value: 1
- Purpose: Defines the integration scheme for propagation of electronic amplitudes (by solving TD-SE)
- Value type: integer
- Possible values:
0 - Based on Trotter splitting formula
10 - Finite difference approach with 1-st order interpolation of Hamiltonian
11 - Finite difference approach with 2-nd order interpolation of Hamiltonian
2 - Exact analytic formula, based on matrix exponential (eigenvalue problem)
- Default value: 0
- Purpose: Nuclear integration time step (slow) in fs. Needed only to determine how many electronic time steps to perform between consequtive time-steps. Typically this is something like 1 fs.
- Value type: real
- Possible values: any (>)
- Default value: 1.0
- Purpose: Electronic integration time step (fast) in fs
- Value type: real
- Possible values: any (>0)
- Default value: 0.001
- Purpose: Defines the temperature of the system (in K units). It is used in the surface-hopping simulations for calculation of the Boltzmann factors affecting the propbability of going to the higher-energy states.
- Value type: real
- Possible values: any (>0)
- Default value: 300.0
- Purpose: defines if the alpha and beta orbitals are coupled
- Value type: integer
- Possible values:
- Default value: 0
0 - in this case the electrons are considered as quasiparticles with spin (alpha and beta, see Figure 1a). This leads to a more detailed structure of the basis states - e.g. states 1 and 2 are distinct, as well as 3 and 4. The alpha and beta orbitals are not coupled, thus transitions 1<-->2, 3<-->4 or 0<-->2 are not allowed, while transitions 0<-->1, 1<-->3 are possible. To developers: the current algorithm, which determines if two states are coupled, is based on the number of occupied orbitals by which two determinants are different. Thus, internally the states 1 and 2 are formally coupled as well as 0 and 2 or 3 and 4. The alp_bet flag set to 0 will simply nullify the NACs (transition dipole moments) for such elements (in KS orbital basis), so effectively the mentioned states will be decoupled.
1 - in this case the electrons are considered as spinless quasiparticles (Figure 1b). This results in the states, defined purely on the spatial parts of the KS orbitals, so no spin-multiplicity is considered and there are no restrictions on possible transitions is due to spin component. The possibility of transitions is solely defined by the occupation numbers of the orbitals. In such an approach states 1 and 2 are equivalent, as well as 3 and 4. The transitions 0<-->(1,2) or (1,2)<-->(3,4) are allowed, while 0<-->(3,4) are not.
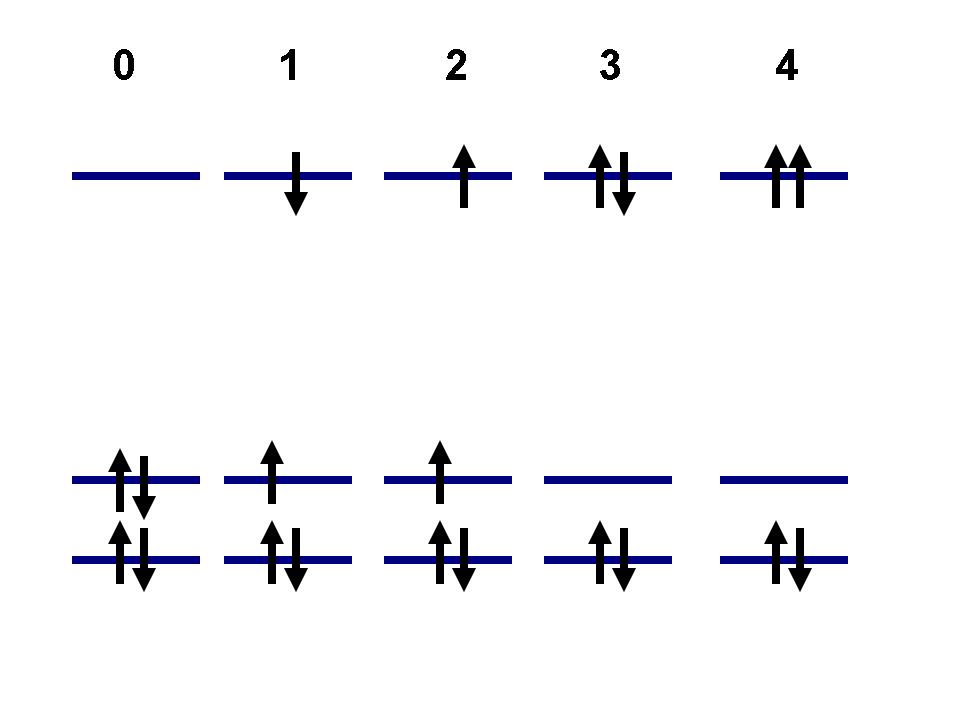
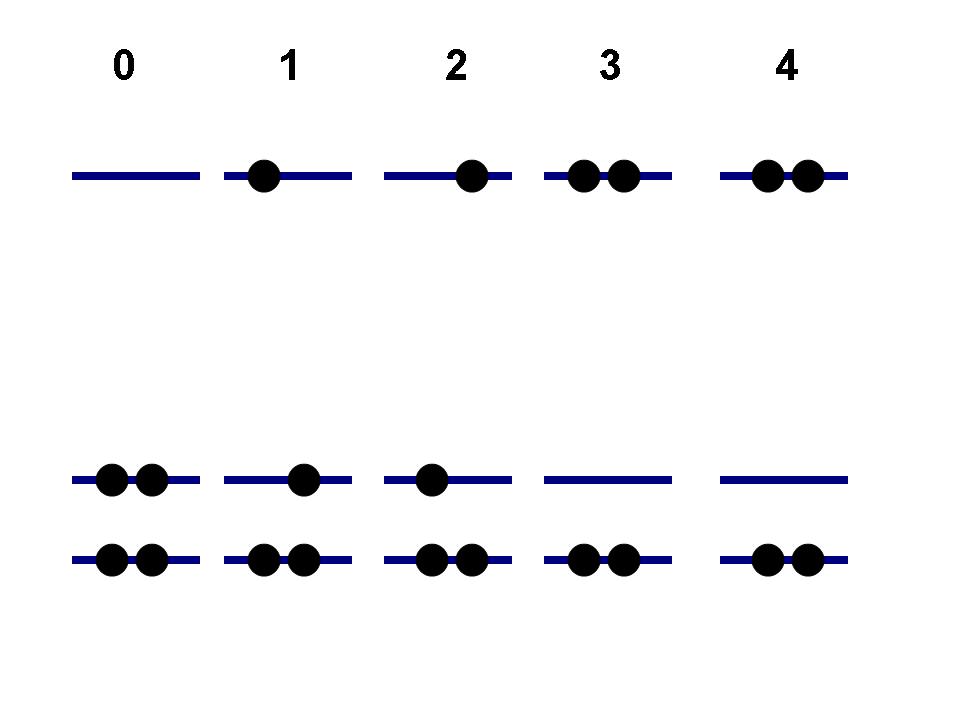
Figure 1. Possible ways to treat coupling between orbitals
- Purpose: Printing some extra information. Carefully - the output may be very massive
- Value type: integer
- Possible values:
0 - do not output extra information
1 - print debug (extra) information
2 - print even more information
...
- Default value: 0
- Purpose: Defines if the decoherence effects should be taken into account and how they should be treated.
- Value type: integer
- Possible values:
0 - no decoherence effects are included
1 - include the decoherence via DISH. In this case the dephasing (decoherence) functions will be computed for each pair of states and for each initial condition. The decoherence functions will be fitted to gaussian curve to determine the dephasing rates. The computed and fitted dephasing functions and the cumulants will be written in the files (located in scratch_dir - see corresponding section). The matrices of dephasing rates will also be written in that directory - one for each initial condition. These files will be re-opened during the main DISH execution cycle. Note: the number of files created is quite large (~Nbas x Nbas x Nicond, where Nbas - the number of basis states Nicond - the number of initial conditions), so be careful (in later version i'll try to fix this)
- Default value: 0
- Purpose: defines the type of fitting to be used in fitting the dephasing functions
- Value type: integer
- Possible values:
0 - y = ax, where P = log(D) and x = t2
1 - y = ax + b, where P = log(D) and x = t2
- Default value: 0